2023年2月28日
是第16个“国际罕见病日”
今年的主题是“点亮你的生命色彩”
罕见病并不罕见
让罕见病被看见

16岁,原本是最灿烂的青春期,但对于高中女孩小李(化名)来说,一丝暗淡却一直伴随着她的成长。
从5岁起,她的运动能力就明显比同龄人要差,甚至无法正常跑跳。父母为此已带她走遍上海、北京等全国各大医院求医,做了多项检查包括肌肉活检等创伤检查,或许因为当时的医疗条件限制,一直没有找到明确的病因。

随着年龄增长,小李的症状越来越重,父母也愈发心疼。家里就这么一个宝贝女儿,就算只有一线希望,父母也始终不愿意放弃。抱着试一试的态度,2020年,父亲带小李来到医院就诊,接诊医生了解情况后,推荐她到神经肌肉专病门诊进一步检查。
神经内科医师仔细评估了小李的情况,结合她的病程及既往的就诊资料,考虑为遗传性疾病的可能性较大。因此给小李做了基因检测,结果证实了最初的判断,小李被确诊为先天性肌无力综合征。


先天性肌无力综合征是一组由不同基因突变造成的常染色体隐性遗传性疾病。
这些突变的基因均可造成神经和肌肉接头的蛋白质功能异常,使神经冲动信号无法正常传递给肌肉引起有力的肌肉收缩。
国外报道,平均发生率约为每100万青少年儿童中9.2人,是当之无愧的罕见病。
和许多遗传性疾病类似
先天性肌无力综合征往往
从儿童、青少年发病
随着年龄的增加
症状逐渐加重至丧失运动功能
甚至危及生命

因为是由于基因突变导致的遗传性疾病,这种病目前基本无法治愈,但可以用药物控制症状。
先天性肌无力综合征有不同的分型,小李姑娘的症状是由于神经肌肉接头处AGRN基因突变所致,极其罕见,可以说是罕见病中的罕见病。夏萍医师通过文献查阅欣喜地发现,国内外有数例报道溴吡斯的明及沙丁胺醇对这一型可能有效。
考虑到药物的副作用,医生先给小李处方了溴吡斯的明,一周后发现效果并不明显,且症状略有加重。果断停用并处方了沙丁胺醇并进行严密的随访。
医师说
2周以后,小李姑娘的父亲激动地发来了信息,女儿肌无力症状有了显著的改善,据他估计大概改善了70%。此时的我内心也是无比激动。我和小李父亲互相加了微信,并告诉他平时如何去关注小李用药疗效、不良反应及如何锻炼等等。
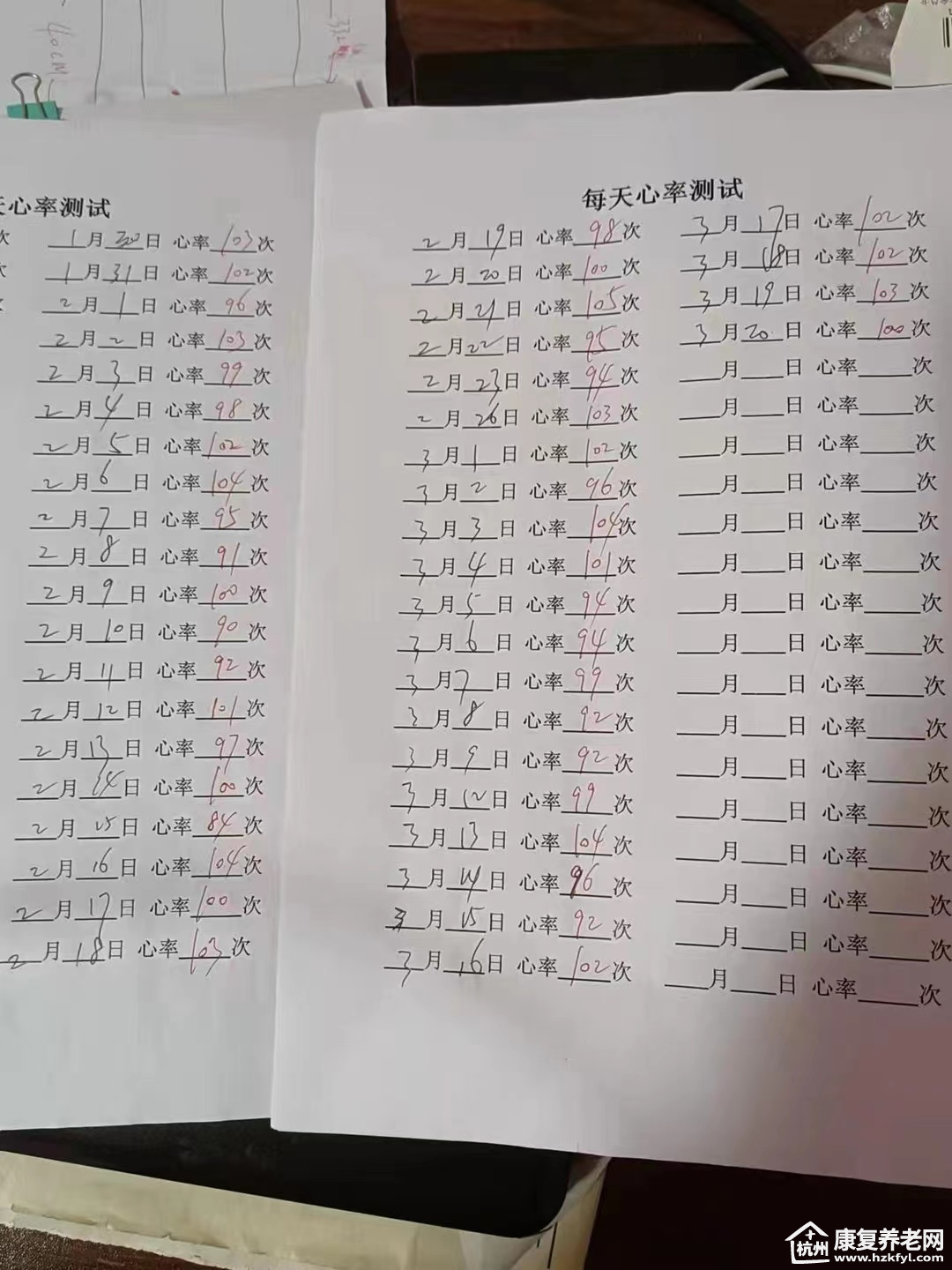
在接下来的整整2年半时间里,小李的父亲每周四都雷打不动发来记录孩子6分钟步行试验的表格,以及运动能力的变化,心率血压记录等,每一张表格,都承载着浓浓的父爱。父亲也定期带小李来医院随访,至今小李对药物依然有良好的反应。




每一位罕见病患者
都不应被放弃

遭遇罕见病,小李无疑是不幸的,但又何其有幸,因为自己的乐观坚强,因为父母的始终坚持,因为医生的认真执着,因为科技的日益发展,得到了最终的诊断;又何其有幸,作为罕见病中的罕见病,又有疗效非常不错,价格又便宜的药物治疗。
2月28日是世界罕见病日,让我们共同关注罕见病。随着医学不断发展,医生掌握诊断和治疗的方法越来越多,神经系统罕见病如DMD肌营养不良,脊髓性肌萎缩症,转甲状腺素蛋白淀粉样变性神经病等都能得到精确的诊断及有效的药物治疗。近年来已不断有罕见病药物上市,并进入国内市场,越来越多的罕见病正在告别“无药可用”的时代。

“每一个小群体,每一位罕见病患者均不应被放弃”,愿越来越多的罕见病患者看到希望的曙光。
以上就是关于“先天性肌无力综合征能治愈吗?【案例分享】”的相关内容,如需咨询了解,请您拨打咨询电话。
杭州余杭区有哪些医养结合的康复医院?
杭州余杭区拥有多家医养结合康复医院,面向老年人群提供集医疗、养老、康复于一体的高...杭州临终关怀医院有哪几家好的医院
杭州临终关怀医院推荐杭州临终关怀医院的选择对于身患重病或即将离世的亲人朋友来说非...杭州临终关怀医院有哪几家医院啊
杭州临终关怀医院推荐——杭州泰康大清谷医院杭州泰康大清谷医院(杭州泰康大清谷安宁...杭州临终关怀医院收费标准最新表
杭州临终关怀医院收费标准最新表杭州泰康大清谷医院(杭州泰康大清谷安宁疗护中心)是...杭州临终关怀医院收费标准最新消息
杭州临终关怀医院收费标准最新消息杭州泰康大清谷医院(杭州泰康大清谷安宁疗护中心)...杭州临终关怀医院有哪些地方好
杭州临终关怀医院推荐——杭州泰康大清谷医院(杭州泰康大清谷安宁疗护中心)咨询电话...杭州临终关怀医院有哪些医院好
杭州临终关怀医院推荐——杭州泰康大清谷医院杭州泰康大清谷医院(杭州泰康大清谷安宁...杭州临终关怀医院地址在哪里
杭州临终关怀医院咨询电话:0571-88261816杭州泰康大清谷医院(杭州泰康...杭州临终关怀医院地址查询
咨询电话:0571-88261816杭州泰康大清谷安宁疗护中心——临终关怀医院杭...杭州临终关怀医院价格查询
杭州临终关怀医院价格查询杭州泰康大清谷医院(杭州泰康大清谷安宁疗护中心)是一家专...